
Muscular Dystrophy
- 30 ago 2023
- 38 min de lectura
Practice Essentials
Muscular dystrophy (MD) is a collective group of inherited noninflammatory but progressive muscle disorders without a central or peripheral nerve abnormality. The disease affects the muscles with definite fiber degeneration but without evidence of morphologic aberrations.
Advances in molecular biology techniques illuminate the genetic basis underlying all types of MD: defects in the genetic code for dystrophin, a 427-kd skeletal muscle protein (Dp427). These defects result in the various manifestations commonly associated with MD, such as weakness and pseudohypertrophy. Dystrophin can also be found in cardiac smooth muscles and in the brain (accounting for the slight mental retardation associated with this disease). [1]
Minor variations notwithstanding, all types of MD have in common progressive muscle weakness that tends to occur in a proximal-to-distal direction, though there are some rare distal myopathies that cause predominantly distal weakness. The decreasing muscle strength in those who are affected may compromise the patient's ambulation potential and, eventually, cardiopulmonary function.
In addition, structural soft-tissue contractures and spinal deformities may develop from poor posturing caused by the progressive muscle weakness and imbalance, all of which can further compromise function and longevity. Equinovarus contractures start as flexible dynamic deformities and advance to rigid contractures. This altered anatomy prevents normal ambulation, proper shoe wear, and transfers (how patients can be picked up to transfer out of their chair).
Once wheelchair-bound, patients with MDs tend to develop worsening contractures, osteopenia (initially, followed by osteoporosis), and rapidly progressive scoliosis. The change in bone mineral density (BMD) can lead to fractures of the long bones or vertebrae, which in turn lead to more immobilization, disuse demineralization, and exacerbation of scoliosis. On average, for each 10° of thoracic scoliosis curvature, forced vital capacity (FVC) decreases by 4%. [2] In a patient with an already weakened cardiopulmonary system, this decrease in FVC could rapidly become fatal.
The goal of orthopedic management is, therefore, to preserve or prolong patients' ambulatory status for as long as possible. This goal can be achieved with soft-tissue releases for contractures, vigilant monitoring of BMD (especially when the patient is on steroid therapy), and treatment of osteoporosis. If patients develop significant scoliosis, which generally occurs after they stop walking, early stabilization of the spine should be considered.
Pathophysiology
Multiple proteins are involved in the complex interactions of the muscle membrane and extracellular environment. For sarcolemmal stability, dystrophin and the dystrophin-associated glycoproteins (DAGs) are important elements. [3, 4]
The dystrophin gene is located on the short arm of chromosome X near the p21 locus and codes for the large protein Dp427, which contains 3685 amino acids. Dystrophin accounts for only approximately 0.002% of the proteins in striated muscle, but it has obvious importance in the maintenance of the muscle's membrane integrity. [2]
Dystrophin aggregates as a homotetramer at the costomeres in skeletal muscles, as well as associates with actin at its N-terminus and the DAG complex at the C-terminus, forming a stable complex that interacts with laminin in the extracellular matrix. Lack of dystrophin leads to cellular instability at these links, with progressive leakage of intracellular components; this results in the high levels of creatine phosphokinase (CPK) noted on routine blood workup of patients with Duchenne MD.
Less active forms of dystrophin may still function as a sarcolemmal anchor, but they may not be as effective a gateway regulator because they allow some leakage of intracellular substance. This is the classic Becker dystrophy. In both Duchenne and Becker MD, the muscle-cell unit gradually dies, and macrophages invade. Although the damage in MD is not reported to be immunologically mediated, class I human leukocyte antigens (HLAs) are found on the membrane of dystrophic muscles; this feature makes these muscles more susceptible to T-cell mediated attacks.
Selective monoclonal antibody hybridization was used to identify cytotoxic T cells as the invading macrophages; complement-activated membrane attack complexes have been identified in dystrophic muscles as well. Over time, the dead muscle shell is replaced by a fibrofatty infiltrate, which clinically appears as pseudohypertrophy of the muscle. The lack of functioning muscle units causes weakness and, eventually, contractures.
Other types of MDs are caused by alterations in the coding of one of the DAG complex proteins. The gene loci coding for each of the DAG complex proteins is located outside the X chromosomes. Gene defects in these protein products also lead to alterations in cellular permeability; however, because of the slightly different mechanism of action and because of the locations of these gene products within the body, there are other associated effects, such as those in ocular and limb-girdle type dystrophies (see the image below).
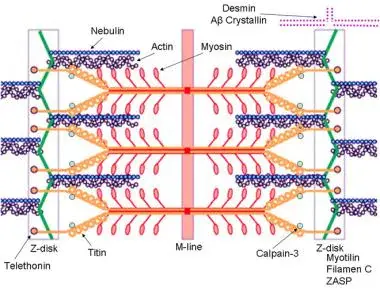
Schematic of the sarcomere with labeled molecular components that are known to cause limb-girdle muscular dystrophy or myofibrillar myopathy. Mutations in actin and nebulin cause the congenital myopathy nemaline rod myopathy, and the mutations in myosin cause familial hypertrophic cardiomyopathy. Image courtesy of Dr F. Schoeni-Affoher, University of Friberg, Switzerland.
Etiology
Classification of types of muscular dystrophy
The etiology of MD is an abnormality in the genetic code for specific muscle proteins. [5] They all are classified according to the clinical phenotype, the pathology, and the mode of inheritance. The inheritance pattern includes the sex-linked, autosomal recessive, and autosomal dominant MDs. Within each group of heritable MDs (see below), several disorders exist. These are characterized by the clinical presentation and pathology.
Heritable MDs include the following:
Sex-linked MDs - Duchenne, Becker, Emery-Dreifuss
Autosomal dominant MDs - Facioscapulohumeral, distal, ocular, oculopharyngeal
Autosomal recessive MD – Limb-girdle form
Genetic defects and dystrophin
In the X-linked forms of MD, such as the Duchenne and Becker dystrophies, the defect is located on the short arm of the X chromosome. [6] Hoffman and coworkers identified the locus of the defect in the Xp21 region, which includes approximately 2 million base pairs. [2] The gene codes for Dp427, which is a component of the cytoskeleton of the cell membrane.
Dystrophin is distributed not only in skeletal muscle but also in smooth and cardiac muscles and in the brain. The large size of the dystrophin gene explains the ease at which spontaneous new mutations can occur, as in Duchenne MD. The large size also allows mistakes in protein synthesis to occur at multiple sites.
Defects that interfere with the translation reading frame or with the promoter sequence that initiates synthesis of dystrophin lead to an unstable, ineffective protein, as in Duchenne MD. Disruption of the translation process further down the sequence leads to production of proteins of lower molecular weight that, although present, are less active and result in the milder variety of Becker MD.
Like Duchenne MD, Emery-Dreifuss MD is a sex-linked recessive disorder, but its defect is localized to the long arm of the X chromosome at the q28 locus. [7] Some authors, however, have cited case reports of similar findings in Emery-Dreifuss that were transmitted in an autosomal dominant pattern. [8] However, this finding is more of an aberration than a normal observation in Emery-Dreifuss MD.
In autosomal recessive conditions such as limb-girdle MD, the genetic defect is localized to the 13q12 locus.
In the autosomal dominant facioscapulohumeral MD, the defect is at the 4q35 locus. In distal MD, it is at the 2q12-14 loci. [9]
Epidemiology
United States statistics
The incidence of MD varies, depending on the specific type of MD under consideration. Duchenne MD is the most common MD and is sex-linked, with an inheritance pattern of 1 case per 3500 live male births. [10, 11] One third of cases occurs as a result of spontaneous new mutations. [12] Becker MD is the second most common form, with an incidence of 1 case per 30,000 live male births. [13] Other types of MD are rare. For example, limb-girdle dystrophy occurs in only 1.3% of patients with MDs.
International statistics
The incidence internationally is similar to that of the US for most of the dystrophies, except for the oculopharyngeal type, which is more common in French Canadians than in other groups. [14] Distal MD tends to occur in Sweden.
Prognosis
Despite modern advances in gene therapy and molecular biology, MD remains incurable. With proper care and attention, patients have a better quality of life than they would otherwise, but most still die by the age of 30 years, usually as a result of cardiopulmonary failure.
Clinical Presentation
History and Physical Examination
Duchenne muscular dystrophy (MD
In Duchenne muscular dystrophy (MD), unless a sibling has been previously affected to warrant a high index of suspicion, no abnormality is noted in the patient at birth, and manifestations of the muscle weakness do not begin until the child begins to walk. Three major time points for patients with Duchenne MD are as follows [15] :
When they begin to walk
When they lose their ability to ambulate
When they die
Phases of Duchenne muscular dystrophy
The child's motor milestones may be at the upper limits of normal, or they may be slightly delayed. Some of the delays may be caused by inherent muscle weakness, but a component may stem from brain involvement.
Although the association of intellectual impairment in MD has long been recognized, it was initially thought to be a result of limited educational opportunities. [16] Psychometric studies have since revealed a definitively lower intelligence quotient (IQ) in patients with Duchenne MD despite equalization of educational opportunities. [17] The average IQ in patients with Duchenne MD is 85 points on the Wechsler Adult Intelligence Scale (WAIS), compared with 105 points in healthy populations. [10, 16, 17, 18]
In addition to mental deficits, another milestone delay is the patient's age at ambulation. Children with Duchenne MD usually do not begin to walk until about age 18 months or later. In the Dubowitz study, [10] 74% of children with Duchenne MD manifested the disease by age 4 years. By age 5 years, awareness increases as the disease is manifested in all affected children when they experience difficulty with school-related activities (eg, getting to the bus, climbing stairs, reciprocal motions during activities).
Other early features include a gait abnormality, which classically is a waddling, wide-based gait with hyperlordosis of the lumbar spine and toe walking. The waddle is due to weakness in the gluteus maximus and gluteus medius muscles and the patient's inability to support a single-leg stance. The child leans the body toward the other side to balance the center of gravity, and the motion is repeated with each step.
Hip extensor weakness also results in a forward tilt of the pelvis, which translates to a hyperlordosis of the spine to maintain posture. The child then walks on tiptoes because it is easier to stay vertical with an equinus foot position than on a flat foot, though no real tendo Achillis contracture exists at this early point.
Gradually, the child is observed to have increasingly noticeable difficulty with step taking. Frequent falls without tripping or stumbling often occur and are described as the feet being swept away from under the child. The child then begins having problems getting up from the sitting or supine position, and he or she can rise to an upright stance only by manifesting the Gowers sign.
The Gowers sign is a classic physical examination finding in MD and results from weakness in the child's proximal hip muscles. To get up from a sitting or supine position, the child must first become prone on the elbows and knees. Next, the knees and elbows are extended to raise the body. Then, the hands and feet are gradually brought together to move the body's center of gravity over the legs. At this point, the child may release one hand at a time and support it on the knee as he or she crawls up their legs to achieve an upright position.
Although the Gowers sign is a classic finding in Duchenne MD, it is by no means pathognomonic; other types of MD and disorders with proximal weakness may also cause this sign.
While still ambulatory, the child may have minimal deformities, including iliopsoas or tendo Achillis tightness. Mild scoliosis may be present if the child has an asymmetrical stance. Upper-extremity involvement rarely occurs in the beginning, although proximal arm muscle weakness may be evident on manual strength testing. When upper-extremity involvement manifests in later stages of Duchenne MD, it is symmetrical and, along with distal weakness, usually follows a rapid worsening of the child's condition toward being wheelchair bound.
The second important phase in Duchenne MD is the loss of ambulation. This usually occurs between the ages of 7 and 13 years, with some patients becoming wheelchair-bound by age 6 years. With early initiation of steroid treatment, prolongation of ambulation potential has been well documented. However, If children with MD are still ambulating well after the age of 13 years, the diagnosis of Duchenne MD should be questioned, because these patients may have Becker MD, the milder form of MD.
In Emery's work, [11] the 50th percentile for loss of ambulation in patients with Duchenne MD was age 8.5 years, with the 95th percentile at 11.9 years and the 99th percentile at 13.2 years. With the child's loss of ambulation, there is usually a rapidly progressive course of muscle or tendon contractures and scoliosis.
Most authors have recommended posterior spinal fusion at 20° when the vital capacity is at its best. [19, 10, 13, 20, 21] However, some reports showed that respiratory function after spinal fusion did not significantly differ. [22, 23, 24, 25, 26] The investigators concluded that respiratory failure resulted from muscle weakness and not the mechanical bellows of the chest cage, as was previously assumed.
Duchenne MD is a terminal disease in which death usually occurs by the third decade of life (mostly from cardiopulmonary compromise), despite steroid treatment. [10] The most common inciting event is a respiratory infection that progresses extremely rapidly despite its initial benign course. The resultant respiratory failure can easily occur from the underlying progressive nocturnal hypoventilation and hypoxia or from an acute cardiac insufficiency.
Additional clinical findings in Duchenne muscular dystrophy
Other clinical findings in Duchenne MD include absent deep tendon reflexes in the upper extremities and patella (though the tendo Achillis reflex remains intact even in the later stages of this disease), pain in the calves with activity (< 30% of patients), pseudohypertrophy of the calf (60%), and macroglossia (30%). Cardiopulmonary involvement is present from the beginning of the disease stages, but the findings are not so clinically obvious. Electrocardiographic (ECG) tracings show right ventricular strain, tall R waves, deep Q waves, and inverted T waves. [27]
Other types of muscular dystrophy
Becker MD is similar to Duchenne MD, but because patients have some measure of functioning dystrophin, the manifestations of Becker MD occur later and are more mild. Patients tend to live past the fourth or fifth decades.
Emery-Dreifuss MD is an uncommon sex-linked dystrophy that presents with early contractures and cardiomyopathy in affected patients; the typical presentation involves tendo Achillis contractures, elbow flexion contractures, neck extension contractures, tightness of the lumbar paravertebral muscles, and cardiac abnormalities. Death may occur in the fourth or fifth decade as a result of first-degree atrioventricular (AV) block, a condition that is usually not present at the initial presentation of this disease.
Autosomal dominant distal MD is a rare form of MD and tends to become apparent in those aged 30-40 years; it is more commonly found in Sweden than in any other country and can cause a mild weakness that affects the arms before the legs.
Autosomal dominant facioscapulohumeral dystrophy causes facial and upper-extremity weakness, and scapulothoracic motion is decreased, with winging of the scapula. This type of dystrophy can occur in both sexes and appear at any age, though it is more common in late adolescence.
Autosomal dominant oculopharyngeal dystrophy appears in those aged 20-30 years. The pharyngeal muscle involvement leads to dysarthria and dysphagia, which may necessitate palliative cricopharyngeal myotomy. The ocular component comprises ptosis, which may not become obvious until the patient's mid life.
None of the autosomal dominant conditions significantly affect longevity.
Complications
Complications of MD usually include the following:
Contractures and early wheelchair dependence, even in patients who develop minor musculoskeletal injuries (eg, ankle sprain) and are immobilized
Weakness - Prolonged immobilization worsens the clinical weakness caused by MD and ultimately renders the patient nonambulatory; this exacerbates any developing contractures and potentiates the development of osteopenia or osteoporosis that can lead to limb and vertebral fractures
Osteoporosis and fractures
Scoliosis
Cardiopulmonary failure
Differential Diagnoses
Workup
Laboratory Studies
A creatine phosphokinase (CPK) determination is the most specific test for muscular dystrophy (MD). Elevated CPK levels are indicative of muscle disease. Because the concentration of CPK is not significant in red blood cells, CPK levels are not affected by hemolysis. CPK is not affected by liver dysfunction, as are the other tested enzymes (eg, transaminases, aldolase, lactate dehydrogenase). High CPK levels represent leakage of the enzyme from the muscle cells only. This change is not exactly correlated with the severity of the disease.
All MDs result in some CPK elevation during the active phase of the disease. The finding of three elevated levels obtained 1 month apart is diagnostic for MD. Early in the disease process, CPK levels are 50-300 times greater than normal levels, but the levels tend to decrease as the muscle mass decreases. The CPK level is highest in Duchenne MD, with less elevation noted in Becker MD.
Enzyme levels that may be elevated but can be altered by liver dysfunction include the following:
Transaminase levels
Lactate dehydrogenase levels
Aldolase levels
The multiplex polymerase chain reaction (PCR) assay may be useful. PCR was developed by Chamberlain et al, [28] who noted that deletions of the dystrophin gene tend to cluster around two hot-spot regions: at exons 3-30 and at exons 44-55. [29] The PCR method rapidly screens for deletions of the dystrophin gene by applying PCR to amplify the DNA in the hot-spot regions and by simultaneously using a number of appropriate primers that flank these hot-spot regions. PCR can be used to detect more than 98% of existing deletions, and it can be performed within 24 hours.
Imaging Studies
Ultrasonography (US) is a relatively noninvasive technique that is used for screening patients with MD; this modality is rapidly replacing electromyography (EMG) in centers that have appropriately trained staff. Even in the early stages of MD, US shows increased echogenicity in the affected muscles, with a corresponding reduction in the underlying bone echo. US has the advantage of noninvasiveness, and it is reliable for continued monitoring of the disease course over time.
Other Tests
Electrocardiography (ECG) is expected to show a right ventricular strain, tall R waves, deep Q waves, and inverted T waves. A cardiologist should be consulted preoperatively because cardiac management may be necessary in the postoperative care of dystrophic patients.
Pulmonary function tests (PFTs), including an analysis of arterial blood gases, and a hematologic workup are necessary as part of the preoperative workup. A pulmonologist may be consulted preoperatively because he or she can be helpful in managing the patient's airway in the postoperative period.
EMG usually demonstrates short-duration, polyphasic, motor-unit action potentials with decreased amplitudes. It should be kept in mind that this finding is common with all myopathic processes and does not specifically identify MDs.
Biopsy
Until the advent of molecular biology techniques, muscle biopsy was the definitive test for diagnosing and confirming muscular disease. Histologic changes depend on the stage of disease and the muscle selected. The optimal site for biopsy is the vastus lateralis, accessed via a small lateral thigh incision.
Histologic Findings
Histologic specimens from muscle biopsy samples obtained early in the development of MD show only variations in muscle fiber sizes with focal areas of degenerating or regenerating fibers. In later stages of MD, the changes are more obvious, with marked variations in muscle fiber sizes, degeneration, and regeneration. Rounded opaque fibers, internal nuclei, splitting of fibers, and a proliferation of connective and adipose tissues are also present. As the disease progresses, fewer and fewer regenerative fibers are seen. In the end phase, the muscle is mostly replaced by adipose tissue, with residual islets of muscle fibers in a sea of fat.
Histochemical staining with the standard adenosine triphosphatase (ATPase) reaction shows a predominance of type I muscle fibers, with loss of clear-cut distinction into the various fiber types. Electron microscopy demonstrates nonspecific degeneration of the fibers, and immunocytochemical techniques show a persistence of fetal and slow myosin in many of these fibers. (See the images below.)
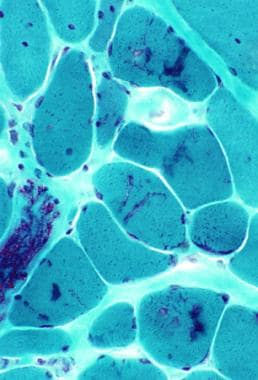
Gomori trichrome–stained section in patient with myofibrillar myopathy. Note the abnormal accumulations of blue-red material in several muscle fibers.
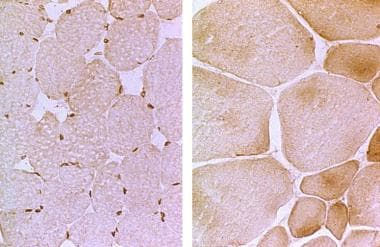
Left: The photomicrograph is a muscle biopsy with normal emerin immunostaining. Right: The micrograph is from a patient with X-linked Emery-Dreifuss muscular dystrophy. Note the absence of nuclear staining as well as the hypertrophied and atrophied muscle fibers.
Treatment & Management
Approach Considerations
The indications for any operative intervention in patients with muscular dystrophy (MD) include making a diagnosis by means of muscle biopsy or prolonging the patient's function and/or ability to ambulate by specific procedures. Other indicated procedures include tendo Achillis and iliopsoas tenotomies for ease of fit into braces, tibialis posterior tendon transfers or tenotomies for more rigid equinovarus deformities of the foot, and segmental spinal stabilization for rapidly developing scoliosis (see Surgical Therapy below).
In patients with MD, some relative contraindications for surgery include obesity, rapidly progressive muscle weakness, poor cardiopulmonary status, and a patient's lack of motivation for participating in postoperative rehabilitation programs.
The ability of advancing technology and molecular biology with fetal blood detection of affected fetuses as early as the first trimester opens the door to many ethical issues. One such issue is whether pregnancy termination should be available as an option when a muscle disease is detected that may be fatal in the third decade of life.
Medical Therapy
Steroids
Since Duchenne's time, multiple drug regimens have been tried in treatment of the muscle weakness. Of all the drugs that have come and gone, the only one with some proven benefit is prednisone. The beneficial effects were initially thought to be mediated through the suppression of cytotoxic T-cell expression from the necrotic muscles.
In the early 1970s, Drachman et al [30] treated 14 boys who had Duchenne MD with steroids and noted some benefits; however, because this was an uncontrolled study, the steroid therapeutic approach did not become a widely accepted treatment protocol.
In 1989, Mendell et al [31] performed a randomized, double-blind, multicenter study of 103 male patients with Duchenne MD who ranged from age 5-15 years. Over a period of 6 months, the patients were given prednisone at a dosage of 1.5 mg/kg/day, prednisone at a dosage of 0.75 mg/kg/day, or placebo. The researchers, who followed the expected course outlined by natural history, noted definite improvement in muscle strength in the steroid-treated boys at 1, 2, and 3 months compared with the control subjects receiving placebo.
The benefit of the dose-dependent steroid in this study, [31] however, was short-lived. The children's gained strength leveled off after the third month, and then they again began to lose strength. In addition, the adverse effects of the higher-dose steroids, such as rapid weight gain, myopathy, osteoporosis, and growth retardation, offset the beneficial effects of temporary minimal increases in strength.
As a result, deflazacort, an oxazoline derivative of prednisolone, is a newer therapeutic drug of choice. [32, 33] Deflazacort reportedly has more bone-sparing and carbohydrate-sparing properties with less weight-gain effects and improves strength and function. Because of the limited side effects and the beneficial properties of muscle sparing and delayed scoliosis progression, deflazacort is being used despite patients' permanent wheelchair status.
Clinical investigations are exploring the possibility of limited courses of steroid bursts (which have shown lasting benefits < 18 months) and other immunosuppressive drugs, such as azathioprine and cyclosporine. Although the glucocorticoid drugs delay the cytotoxic damage of MD to the necrosing muscle cells, these drugs cannot and do not make, or stimulate the synthesis of, the dystrophin and DAG proteins that are deficient, which is the root cause of the disease.
Biologic agents
Eteplirsen
An open-label, phase 2, dose-escalation study evaluated the safety and efficacy of intravenously administered eteplirsen phosphorodiamidate morpholino oligomer (PMO) in patients with Duchenne MD. [34] Using data from 19 ambulant patients aged 5-15 years with amenable deletions in Duchenne MD, the investigators noted that eteplirsen was well tolerated with no serious drug-related adverse events; eteplirsen induced exon 51 skipping in all cohorts and new dystrophin protein expression in a significant dose-dependent, but variable, manner in boys (dose 2 mg/kg) onwards.
Early clinical results of eteplirsen were biochemically promising for dystrophin production without significant adverse effects; however, functional ambulatory changes were not as consistently correlated. [34] Reevaluation of the existing data led to approval of eteplirsen in September 2016.
Golodirsen
Golodirsen, a second antisense oligonucleotide, was approved by the US Food and Drug Administration (FDA) in December 2019. Golodirsen is indicated for treatment of Duchenne MD in patients who have a confirmed mutation of the DMD gene that is amenable to exon 53 skipping. Approval was based on an increase in dystrophin production in skeletal muscle observed in patients who were treated. Mean dystrophin levels increased from 0.1% of normal at baseline to 1.02% of normal by week 48, with a mean change in dystrophin of 0.92% of normal levels; the median change from baseline was 0.88%. [35]
Viltolarsen
Viltolarsen, another morpholino antisense oligomer, was approved by the FDA in August 2020 for Duchenne MD in patients with genetic mutations that are amenable to exon 53 skipping. Approval was based on results from a phase 2 two-period study in patients aged 4 to less than 10 years conducted in North America (N = 16) and a multicenter, open-label study in boys aged 5 to less than 18 years conducted in Japan (N = 16). In the former, 100% of patients who received the recommended dose of 80 mg/kg/wk (N = 8) showed an increase in dystrophin levels after treatment. [36]
Casimersen
In February 2021, the FDA granted accelerated approval to casimersen, an antisense oligonucleotide of the PMO subclass. PMO binds to exon 45 of dystrophin pre-mRNA, resulting in exclusion of this exon during mRNA processing in patients with genetic mutations that are amenable to exon 53 skipping. Casimersen is indicated for Duchenne MD in patients with a confirmed mutation amenable to exon 45 skipping.
The ESSENCE trial, a placebo-controlled confirmatory trial to support the casimersen approval, is ongoing and expected to conclude in 2024. In the study, patients who received casimersen showed a significantly greater increase in dystrophin protein levels from baseline to week 48 of treatment compared to those who received placebo. [37]
Investigational agents
Another potential therapy is creatine monohydrate supplementation. Creatine is a natural compound occurring in meats and is also endogenously produced by the liver and kidneys. Creatine supplementation has been shown to enhance athletic performance of healthy individuals in up to 10%. [38, 39] Studies looking at creatine use in neuromuscular disorders have been popularized since late 1997, with the publication of the first human study by Tarnopolksy et al showing an increase in high-intensity power output with use. [40] Several other human clinical trials of creatine supplementation have been conducted since that time with similar results. [41, 42, 43]
A meta-analysis of all randomized clinical trials using creatine monohydrate supplementation in neuromuscular disorders versus placebo was performed. [44] It found that short- and intermediate-term treatment with 0.03-0.04 g/kg/day of creatine monohydrate supplementation resulted in modest but significant increases in mean maximum voluntary contraction of 9.2 N higher than placebo. There is also an increase in fat-free muscle mass. Globally, 44% of patients felt better in the creatine treated group, compared with 10% in the placebo group.
These effects were reliably seen in patients with dystrophinopathies and type II myotonic myopathy. [44] No consistent changes were noted in patients with type I myotonic dystrophy, and none were noted in those with metabolic myopathies. Although there were no serious side effects noted in most patients, high-dose creatine treatment can impair ADL and increase muscle pain in glycogen storage disease type V (McArdle disease).
Ataluren (formerly known as PTC124; PTC Therapeutics, Inc, South Plainfield, NJ) is an oxadiazole compound that, when taken orally, can override nonsense stop translation signals induced by the dystrophin gene mutation; the protein produced is thus the full-length protein. [45] In Europe, it is used to treat Duchenne MD patients who have a nonsense mutation in the dystrophin gene, can walk, and are older than 2 years. [46] It is not approved by the FDA for this purpose.
Gene therapy
In June 2023, the first gene therapy, delandistrogene moxeparvovec (Elevidys), gained accelerated approval by the FDA for ambulant Duchenne MD pediatric patients aged 4-5 years with a confirmed mutation in the DMD gene. It is administered as a one-time infusion designed to treat the underlying genetic cause of Duchenne MD: mutations in the dystrophin gene that result in the lack of dystrophin protein.
Approval was supported by the ENDEAVOR trial. [47] Exploratory subgroup analyses showed that for participants aged 4-5 years, the least squares mean changes in North Star Ambulatory Assessment (NSAA) total score from baseline to Week 48 were 4.3 (0.7) points for the delandistrogene moxeparvovec group, and 1.9 (0.7) points for the placebo group, demonstrating a numerical advantage for delandistrogene moxeparvovec. [48]
The open-label multicenter EMBARK trial will serve as the confirmatory trial required for the accelerated approval. [49]
Early gene therapy investigations
Intense investigation of somatic gene therapy began in the 1990s. Healthy immature myoblasts were introduced into the diseased muscles, which then fused and stimulated production of enough dystrophin to reverse the degeneration that occurs in the affected muscles. [50]
In earlier states of research, somatic gene therapy was achieved successfully in the X-linked muscular dystrophic mouse (murine MDX) model with the fusion of the donor and host muscle cells, which expressed some dystrophin, but the benefit was not thought to translate into human males. [50] The mice could not demonstrate muscle strength, and the laboratory-raised mice were not able to mount a rejection response such as might occur in humans. Other investigations have been conducted on the canine MDX model, which more closely approximates the human condition. [51, 52]
Human trials of gene therapy began in 1990, with an uncontrolled trial of eight patients who were injected with myoblasts from family donors. [53] Strength testing and staining for dystrophin was performed after several months. Early results demonstrated no improvement in patients' muscle strength or dystrophin staining. Later studies showed an increase in the expression of dystrophin proteins. However, the clinical results remained unchanged. These preliminary results, though disappointing, did not dampen the promise of gene therapy. Most supporters believed that these failures were merely the result of a lack of expertise, as with once-novel techniques such as organ transplantation.
Other molecular approaches to therapy include recombinant versions of the dystrophin gene using viral or nonviral vectors and antisense oligonucleotides. [54, 55] In the viral vector therapeutic approach, adenosine-associated virus led the way. [56] In nonviral gene therapy, plasmid-mediated gene delivery, antisense-mediated exon skipping, and oligonucleotide-mediated gene editing moved from successful trials in the laboratory to the clinic. In approximately 10-20% of the preclinical cases, [55] it was possible to chemically persuade the translational machinery to read through a premature stop codon, as noted with the dystrophin mutation, leading to production of a more functional full-length protein.
Surgical Therapy
The orthopedic problems in children with MD are as follows:
Progressive weakness with loss of ambulatory status
Soft-tissue contractures
Spinal deformities
The role of the orthopedic surgeon is to correct the deformities and to help maintain the dystrophic child's ambulatory status for as long as possible, usually 1-3.5 years. [57, 58] The modalities available to obtain these goals were well outlined by Drennan [59] and include the following:
Functional testing
Physical therapy
Use of orthoses
Fracture management
Soft-tissue, bone, and spinal surgeries
Use of a wheelchair when indicated
Genetic and/or psychological testing
Nonoperative measures
Functional testing, as its name implies, refers to frequent evaluation of the involved muscle's range of motion and strength. This modality provides therapists with goals for patients' individualized therapy programs. Regular therapy sessions are necessary because the therapist also works with patients for gait training and transfer techniques. The use of all adaptive equipment is considered necessary by the orthopedist to maintain the patient's ambulatory status.
Drennan also recommended that a home program be taught to dystrophic patients, with stretching exercises for the lower extremities performed twice a day on a firm surface to minimize contractures. [59] Occasionally, serial casting may be necessary to manage significant flexion contractures at the knee or equinus contracture at the ankle.
The goal for patients with MD is continued mobility despite the use of a cast to prevent rapid loss of strength and bone mineral density. Even with initial loss of muscle strength for weightbearing, flexible soft-tissue and rigid ankle-foot orthosis (AFO) or ischial supportive knee-ankle-foot orthosis (KAFO) can help the patient maintain standing balance for additional months to years.
Operative approaches to contractures and deformities
Significant upper-extremity contractures rarely occur in patients with MD. Occasionally, tightness of the long flexors may become problematic with hand function in operating an automatic wheelchair, but historically this has been treated with a nighttime orthosis.
In patients with MD, lower-extremity contractures start with equinus deformities. Initially, the contractures are supple and can occur in children as young as 6 years. Patients may be treated with various types of procedures to lengthen the tendo Achillis, including Vulpius tendon lengthening (which avoids overlengthening the already weakened muscle), percutaneous Hoke-Miller triple cut (which limits the incision and, thus, the postoperative immobilization), Warren-White lengthening, or standard z-lengthening.
Occasionally, when an associated varus deformity is present as a result of overpull of the unaffected tibialis posterior muscle, a posterior tibial tendon transfer through the interosseous membrane or a split-posterior tibial tendon transfer may also be indicated. In cases of severe contractures at the foot and ankle, posterior tibial lengthening or tenotomy may be necessary to achieve a plantigrade position.
Hip and knee contractures develop later in MD. Once patients become wheelchair-bound, hip and knee flexion contractures are more rapidly progressive. The abduction contracture was initially thought to be useful in obtaining stability with a wider base gait, but it also makes it difficult for patients to fit in standard wheelchairs or to be comfortable in bed. Various tenotomies are available, including the Yount (resection of the iliotibial band [ITB] distally at the knee), [60] modified Souter-Strathclyde (resection of the ITB proximally), and complete resection of the ITB from the hip to the knee.
Transfer of the iliopsoas has also been tried with limited success; this is no longer a procedure of choice in patients with MD. Posterior capsulotomy of the knee can allow for maintenance of flexible extremities for bracing, although this is not routinely performed. KAFOs with locked knees may extend the ambulatory status of weakened patients by 1 to 3 years. [58, 13] The locked knees, however, are not well tolerated, as they cause children to feel as if they are going to fall. This is a worse condition than buckling because of weakness at the quadriceps. Postoperatively, all patients are given some type of orthosis.
The surgical approaches to contractures in dystrophic patients, especially those with Duchenne MD, can be summarized into the following three broad categories:
Ambulatory
Rehabilitative
Palliative
Within the ambulatory group, the approach can be aggressive, so that all contractures are addressed at the start, before patients lose ambulatory status or within the first month of their losing ambulatory status. The rehabilitative approach indicates that surgery is used only to correct deformities that may limit physical therapy and orthosis wear. The palliative approach, as its name implies, is used to treat only problems of immediate concern for the patient's comfort, such as difficulty with shoe wearing, ulcerations, and problematic positioning in wheelchairs. Early aggressive surgical releases can prolong the patient's ambulatory status as long as 3.5 years. [57, 58]
Scoliosis is another common problem in patients with muscular dystrophy. It is more common in Duchenne MD than in other forms, with an incidence of 75-90%. [57, 58, 19] The scoliosis develops early and tends to be rapidly progressive, especially when patients become nonambulatory. The curve is usually thoracolumbar or lumbar with associated pelvic obliquity, thoracic kyphosis, and lumbar hyperlordosis. The abnormal sagittal alignment may cause problems with seating systems, even modified systems, and the rapid progression of the scoliosis requires frequent wheelchair adjustments. Braces are not effective in progressive paralytic or neuromuscular curves, and surgery is often indicated. [61]
Advances in pulmonary care and cardiac drugs may negate the absolute need for scoliosis surgery in Duchenne MD, allowing patients to live into adulthood. A 10-year retrospective study showed that 44 of 123 nonambulatory patients aged 17 or older were managed satisfactorily without surgery. [23] Although this is only a single report, it gives much hope for many dystrophic patients with scoliosis.
The technique of choice for scoliosis when the curve measures 20° or more in patients who are nonambulatory is a posterior spinal fusion from T2 to the sacrum. The indication for earlier operative stabilization of the spine in these patients is due to the rapidly deteriorating cardiopulmonary function. [26] The FVC is at its maximum in children younger than 10 years. After this point, the FVC rapidly declines, and anesthetic complications rise dramatically in those patients with an FVC less than 30%.
Spinal fusion is extended to the pelvis, with complete obliteration of the facet joints to ensure arthrodesis. The instrumentation used has often consisted of a Luque rod with segmental sublaminar wires to the L5 level, with bone arthrodesis extending into the sacrum. Currently, however, this method is being employed less frequently; most spine surgeons are now using pedicle screws with extension of the instrumentation to the pelvis. [62, 63]
Occasionally, instrumentation and fusion are extended only to L5 because of the diffuse osteopenia in the sacrum, early surgery and low magnitude curves, or because of the possible complications of instrument failure. The literature has cited a high incidence of failure and revision with fusion short of the sacrum; therefore, spinal fusions in patients with MD are extended to the pelvis, especially if done after the curve magnitude is over 30º. [64, 65, 66]
Preoperative considerations
Before any operative is undertaken, the patient's overall status of any patient must be considered; this is of particular importance in patients with muscle weakness, such as those with MD. For example, posterior spinal fusion to the pelvis straightens the scoliosis and allows better upright sitting balance. However, in patients with low vital capacity (< 30%), the risks of pulmonary complications are much higher, and these risks may tip the scale in favor of not operating on the scoliosis.
Other examples include equinus contractures in patients who are very weak; tendon lengthening itself is necessarily a weakening procedure on the involved muscle. If the patient has to maintain a rigid equinus foot position for stability of gait and the tendon is lengthened by surgery, the patient will not be able to ambulate.
Preoperatively, patients should undergo a detailed cardiac assessment, a pulmonary evaluation with pulmonary function tests (including arterial blood gas analysis), and a hematologic workup. Because of potential cardiomyopathy, intraoperative monitoring is an essential component of administering anesthetics.
Intraoperative blood loss is usually substantial in patients with MD as a result of their muscle dysfunction, which causes ineffective vessel constriction. Another potential complication of anesthesia is malignant hyperthermia, which is more common in patients with muscle diseases than in patients with other disease entities; this risk is diminished with the use of nitrous oxide, intravenous narcotics, sedatives, and nondepolarizing muscle relaxants.
After scoliosis surgery, patients may need additional pulmonary support and an extended stay in the intensive care unit (ICU). Preoperative tracheostomy is usually not any more effective in early mobilization of dystrophic patients; if necessary, this procedure is performed only after the patient's condition has been stabilized and after a mold has been obtained for a hard brace with chest and abdominal cutouts.
Management of Osteoporosis and Fractures
Whereas the presence of bone fragility due to limited mobility, decreased vitamin D absorption, and lack of exercise in the sun is historically well attested in MD, the common practice of routinely using steroids for mitigation of MD weakness has led to increased concerns regarding steroid-exacerbated osteoporotic long bone and vertebral fractures.
In 2014, Tian et al retrospectively reviewed a cohort of 408 males with Duchenne MD and found an age-dependent increased prevalence in total as well as vertebral compression fractures. [67] Singh et al further confirmed this finding in their study of 49 patients: 26 of the 49 had vertebral fractures, with 50% occurring within 5 years of steroid therapy, 69% within 7 years of steroid use, and 100% after 9 years of steroid treatment. [68] The investigators concluded that there is a high risk of vertebral fractures associated with the duration of deflazacort use, regardless of the age at which steroid therapy is initiated.
Vertebral fractures tend to occur via a bimodal distribution among children with osteoporosis, with the incidence of symptomatic fractures ranging from 32% to 40%. This is important in that previous vertebral fractures, regardless of symptomology, are a powerful predictor of future fractures and guide recommendations for osteoporosis treatment.
In a very thorough review, Buckner et al detailed the recommendations for screening and treatment of osteoporosis induced by limited mobility and chronic steroid use. [69] An initial thorough dietary screen for vitamin D and calcium intake is done at the initial visit and at regular intervals thereafter. A fracture screen is also performed. Active vitamin D (25-hydroxyvitamin D) levels can be checked by means of a blood test. The Endocrine Society defines the following categories for vitamin D levels:
Vitamin D deficiency - < 20 ng/mL
Insufficient vitamin D - < 30 ng/mL but >20 ng/mL
Sufficient vitamin D - >30 ng/mL
A baseline dual energy x-ray absorptiometry (DXA) scan is performed when steroid therapy is started and is repeated every 1-2 years while therapy continues. The scan is done with height-adjusted Z-scores, which yield more consistent results for pediatric MD patients with shorter stature. With the onset of back pain, a lateral radiograph is obtained as a baseline; lateral spine x-rays involve more radiation than the DXA scan.
If the patient has low Z-scores, a history of previous fractures, or both, treatment of the osteoporosis is indicated. Therapeutic options include the following:
Calcium and vitamin D supplementation
“Vibration” therapy (which has been shown to be more effective in cerebral palsy patients than other categories of neurologic patients)
Bisphosphonates - Bisphosphonates typically used in pediatric patients include oral alendronate, intravenous pamidronate, and zoledronic acid; these agents have been shown to improve bone mineral density (BMD) and Z-scores as well as ease fracture-induced back pain
Multiple studies are currently being conducted to further elucidate clear recommendations for the diagnosis and treatment of steroid-induced osteoporotic fractures of the spine and long bones in patients with MD.
Medication
Medication Summary
Several agents have shown promise in management of Duchenne muscular dystrophy (MD). The mainstay of therapy has been the steroids; however, the lack of prolonged efficacy and the attendant adverse effects of higher-dose steroids limit their use. Deflazacort, an oxazoline derivative of prednisolone, may offer therapeutic efficacy with fewer adverse effects.
The first gene therapy, delandistrogene moxeparvovec (Elevidys), gained accelerated FDA approval in June 2023 for the treatment of Duchenne MD in ambulant pediatric patients aged 4-5 years with a confirmed mutation in the DMD gene.
The approval of eteplirsen, the first phosphorodiamidate morphino oligomer (PMO), provides an addition to the therapeutic armamentarium; however, continued approval of eteplirsen is contingent on postmarketing performance data. A second such agent, golodirsen, was subsequently approved. Other morpholino antisense oligomers, viltolarsen and casimersen, have also been approved by the FDA for use in the United States. [34, 35, 36, 37]
Corticosteroids Class Summary Glucocorticoid drugs delay the cytotoxic damage of MD to the necrosing muscle cells. Prednisone (Deltasone, Prednisone Intensol, Rayos)
Prednisone exhibits beneficial effects in treatment of Duchenne MD in dosages ranging from 0.75 mg/kg/day to 1.5 mg/kg/day. Therapeutic effects are believed to be mediated through suppression of cytotoxic T-cell expression from necrotic muscles. Response to therapy may not last beyond several months, and adverse effects (eg, rapid weight gain, osteoporosis, myopathy, and growth retardation) limit its use. Deflazacort (Emflaza)
Deflazacort, an oxazoline derivative of prednisolone, represents one of the newer corticosteroids for Duchenne MD. The FDA-approved dose is 0.9 mg/kg/day. It elicits a therapeutic response similar to that of prednisone.
Morpholino Antisense Oligomers, Neurology Class Summary These agents bind to various exons (eg, 45, 51, 53) of dystrophin pre-mRNA, resulting in exclusion of the particular exon (ie, skipping) during mRNA processing. Exon skipping is intended to allow production of an internally truncated dystrophin protein in patients with genetic mutations that are amenable to specific exon skipping. Eteplirsen (Exondys 51)
Eteplirsen is a phosphorodiamidate morphino oligomer (PMO), the first of its class. It is indicated for Duchenne MD in patients who have a confirmed mutation of the DMD gene that is amenable to exon 51 skipping. It is administered as a once-weekly IV infusion. Golodirsen (Vyondys 53)
A second PMO was approved for Duchenne MD in patients who have a confirmed mutation of the dystrophin gene that is amenable to exon 53 skipping. Viltolarsen (Viltepso) Antisense oligonucleotide of the phosphorodiamidate morpholino oligomer (PMO) subclass that binds to exon 53 of dystrophin pre-mRNA. This results in exclusion of this exon during mRNA processing in patients with genetic mutations that are amenable to exon 53 skipping. It is indicated for Duchenne muscular dystrophy (DMD) in patients with a confirmed DMD gene mutation that is amenable to exon 53 skipping. Casimersen (Amondys 45)
Casimersen is an antisense oligonucleotide of the phosphorodiamidate morpholino oligomer (PMO) subclass. PMO binds to exon 45 of dystrophin pre-mRNA, resulting in exclusion of this exon during mRNA processing in patients with genetic mutations that are amenable to exon 53 skipping. Casimersen is indicated for Duchenne muscular dystrophy (DMD) in patients with a confirmed mutation amenable to exon 45 skipping.
Gene Therapies, Neurologics Class Summary Gene therapies are designed to treat the underlying genetic cause of Duchenne MD: mutations in the dystrophin gene that result in the lack of dystrophin protein. Delandistrogene moxeparvovec (Elevidys)
One-time gene therapy indicated for Duchenne MD in ambulant pediatric patients aged 4-5 years with a confirmed mutation in the DMD gene.
References
Yanagisawa A, Bouchet C, Quijano-Roy S, Vuillaumier-Barrot S, Clarke N, Odent S, et al. POMT2 intragenic deletions and splicing abnormalities causing congenital muscular dystrophy with mental retardation. Eur J Med Genet. 2009 Jul-Aug. 52 (4):201-6. [QxMD MEDLINE Link].
Hoffman EP, Brown RH Jr, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987 Dec 24. 51 (6):919-28. [QxMD MEDLINE Link].
Waite A, Tinsley CL, Locke M, Blake DJ. The neurobiology of the dystrophin-associated glycoprotein complex. Ann Med. 2009. 41 (5):344-59. [QxMD MEDLINE Link].
Banks GB, Chamberlain JS, Froehner SC. Truncated dystrophins can influence neuromuscular synapse structure. Mol Cell Neurosci. 2009 Apr. 40 (4):433-41. [QxMD MEDLINE Link].
Bushby K. Genetics and the muscular dystrophies. Dev Med Child Neurol. 2000 Nov. 42 (11):780-4. [QxMD MEDLINE Link].
González-Herrera L, Gamas-Trujillo PA, García-Escalante MG, Castillo-Zapata I, Pinto-Escalante D. [Identifying deletions in the dystrophin gene and detecting carriers in families with Duchenne's/Becker's muscular dystrophy]. Rev Neurol. 2009 Jan 16-31. 48 (2):66-70. [QxMD MEDLINE Link].
Dickey RP, Ziter FA, Smith RA. Emery-Dreifuss muscular dystrophy. J Pediatr. 1984 Apr. 104 (4):555-9. [QxMD MEDLINE Link].
Miller RG, Layzer RB, Mellenthin MA, Golabi M, Francoz RA, Mall JC. Emery-Dreifuss muscular dystrophy with autosomal dominant transmission. Neurology. 1985 Aug. 35 (8):1230-3. [QxMD MEDLINE Link].
Dobrowski JM, Zajtchuk JT, LaPiana FG, Hensley SD Jr. Oculopharyngeal muscular dystrophy: clinical and histopathologic correlations. Otolaryngol Head Neck Surg. 1986 Sep. 95 (2):131-42. [QxMD MEDLINE Link].
Dubowitz V. Muscle Disorders in Childhood. 2nd ed. Philadelphia: WB Saunders; 1995. 34-132.
Emery AEH, Muntoni F, Quinlivan R. Duchenne Muscular Dystrophy (Oxford Monographs on Medical Genetics). 4th ed. Oxford, UK: Oxford University Press; 2015.
Emery AE. Population frequencies of inherited neuromuscular diseases--a world survey. Neuromuscul Disord. 1991. 1 (1):19-29. [QxMD MEDLINE Link].
Shapiro F, Specht L. The diagnosis and orthopaedic treatment of inherited muscular diseases of childhood. J Bone Joint Surg Am. 1993 Mar. 75 (3):439-54. [QxMD MEDLINE Link].
Pratt MF, Meyers PK. Oculopharyngeal muscular dystrophy: recent ultrastructural evidence for mitochondrial abnormalities. Laryngoscope. 1986 Apr. 96 (4):368-73. [QxMD MEDLINE Link].
Donders J, Taneja C. Neurobehavioral characteristics of children with Duchenne muscular dystrophy. Child Neuropsychol. 2009 May. 15 (3):295-304. [QxMD MEDLINE Link].
Prosser EJ, Murphy EG, Thompson MW. Intelligence and the gene for Duchenne muscular dystrophy. Arch Dis Child. 1969 Apr. 44 (234):221-30. [QxMD MEDLINE Link].
Leibowitz D, Dubowitz V. Intellect and behaviour in Duchenne muscular dystrophy. Dev Med Child Neurol. 1981 Oct. 23 (5):577-90. [QxMD MEDLINE Link].
Pane M, Lombardo ME, Alfieri P, D'Amico A, Bianco F, Vasco G, et al. Attention deficit hyperactivity disorder and cognitive function in Duchenne muscular dystrophy: phenotype-genotype correlation. J Pediatr. 2012 Oct. 161 (4):705-9.e1. [QxMD MEDLINE Link].
Kay RM, Swaroop VT. Other neuromuscular disorders. Weinstein SL, Flynn JM, Crawford HA, eds. Lovell and Winter's Pediatric Orthopaedics. 8th ed. Philadelphia: Wolters Kluwer; 2021. Vol 1: 591-658.
Sussman MD. Advantage of early spinal stabilization and fusion in patients with Duchenne muscular dystrophy. J Pediatr Orthop. 1984 Sep. 4 (5):532-7. [QxMD MEDLINE Link].
Weimann RL, Gibson DA, Moseley CF, Jones DC. Surgical stabilization of the spine in Duchenne muscular dystrophy. Spine (Phila Pa 1976). 1983 Oct. 8 (7):776-80. [QxMD MEDLINE Link].
Almenrader N, Patel D. Spinal fusion surgery in children with non-idiopathic scoliosis: is there a need for routine postoperative ventilation?. Br J Anaesth. 2006 Dec. 97 (6):851-7. [QxMD MEDLINE Link].
Kinali M, Messina S, Mercuri E, Lehovsky J, Edge G, Manzur AY, et al. Management of scoliosis in Duchenne muscular dystrophy: a large 10-year retrospective study. Dev Med Child Neurol. 2006 Jun. 48 (6):513-8. [QxMD MEDLINE Link].
Birnkrant DJ. New challenges in the management of prolonged survivors of pediatric neuromuscular diseases: a pulmonologist's perspective. Pediatr Pulmonol. 2006 Dec. 41 (12):1113-7. [Full Text].
Miller RG, Chalmers AC, Dao H, Filler-Katz A, Holman D, Bost F. The effect of spine fusion on respiratory function in Duchenne muscular dystrophy. Neurology. 1991 Jan. 41 (1):38-40. [QxMD MEDLINE Link].
Miller F, Moseley CF, Koreska J, Levison H. Pulmonary function and scoliosis in Duchenne dystrophy. J Pediatr Orthop. 1988 Mar-Apr. 8 (2):133-7. [QxMD MEDLINE Link].
Thrush PT, Allen HD, Viollet L, Mendell JR. Re-examination of the electrocardiogram in boys with Duchenne muscular dystrophy and correlation with its dilated cardiomyopathy. Am J Cardiol. 2009 Jan 15. 103 (2):262-5. [QxMD MEDLINE Link].
Chamberlain JS, Gibbs RA, Ranier JE, Nguyen PN, Caskey CT. Deletion screening of the Duchenne muscular dystrophy locus via multiplex DNA amplification. Nucleic Acids Res. 1988 Dec 9. 16 (23):11141-56. [QxMD MEDLINE Link]. [Full Text].
Miyazaki D, Yoshida K, Fukushima K, Nakamura A, Suzuki K, Sato T, et al. Characterization of deletion breakpoints in patients with dystrophinopathy carrying a deletion of exons 45-55 of the Duchenne muscular dystrophy (DMD) gene. J Hum Genet. 2009 Feb. 54 (2):127-30. [QxMD MEDLINE Link].
Drachman DB, Toyka KV, Myer E. Prednisone in Duchenne muscular dystrophy. Lancet. 1974 Dec 14. 2 (7894):1409-12. [QxMD MEDLINE Link].
Mendell JR, Moxley RT, Griggs RC, Brooke MH, Fenichel GM, Miller JP, et al. Randomized, double-blind six-month trial of prednisone in Duchenne's muscular dystrophy. N Engl J Med. 1989 Jun 15. 320 (24):1592-7. [QxMD MEDLINE Link].
McAdam LC, Mayo AL, Alman BA, Biggar WD. The Canadian experience with long-term deflazacort treatment in Duchenne muscular dystrophy. Acta Myol. 2012 May. 31 (1):16-20. [QxMD MEDLINE Link].
Lebel DE, Corston JA, McAdam LC, Biggar WD, Alman BA. Glucocorticoid treatment for the prevention of scoliosis in children with Duchenne muscular dystrophy: long-term follow-up. J Bone Joint Surg Am. 2013 Jun 19. 95 (12):1057-61. [QxMD MEDLINE Link].
Cirak S, Arechavala-Gomeza V, Guglieri M, Feng L, Torelli S, Anthony K, et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet. 2011 Aug 13. 378 (9791):595-605. [QxMD MEDLINE Link].
Vyondys 53 (golodirsen) [package insert]. Cambridge, MA: Sarepta Therapeutics. December 2019. Available at [Full Text].
Viltepso (viltolarsen) [package insert]. Paramus, NJ: NS Pharma, Inc. August 2020. Available at [Full Text].
Study of SRP-4045 and SRP-4053 in DMD Patients (ESSENCE). Clinicaltrials.gov. Available at https://www.clinicaltrials.gov/ct2/show/NCT02500381. January 19, 2023; Accessed: January 24, 2023.
Casey A, Constantin-Teodosiu D, Howell S, Hultman E, Greenhaff PL. Creatine ingestion favorably affects performance and muscle metabolism during maximal exercise in humans. Am J Physiol. 1996 Jul. 271 (1 Pt 1):E31-7. [QxMD MEDLINE Link].
Tarnopolsky MA, MacLennan DP. Creatine monohydrate supplementation enhances high-intensity exercise performance in males and females. Int J Sport Nutr Exerc Metab. 2000 Dec. 10 (4):452-63. [QxMD MEDLINE Link].
Tarnopolsky MA, Roy BD, MacDonald JR. A randomized, controlled trial of creatine monohydrate in patients with mitochondrial cytopathies. Muscle Nerve. 1997 Dec. 20 (12):1502-9. [QxMD MEDLINE Link].
Chung YL, Alexanderson H, Pipitone N, Morrison C, Dastmalchi M, Ståhl-Hallengren C, et al. Creatine supplements in patients with idiopathic inflammatory myopathies who are clinically weak after conventional pharmacologic treatment: Six-month, double-blind, randomized, placebo-controlled trial. Arthritis Rheum. 2007 May 15. 57 (4):694-702. [QxMD MEDLINE Link].
Klopstock T, Querner V, Schmidt F, Gekeler F, Walter M, Hartard M, et al. A placebo-controlled crossover trial of creatine in mitochondrial diseases. Neurology. 2000 Dec 12. 55 (11):1748-51. [QxMD MEDLINE Link].
Louis M, Lebacq J, Poortmans JR, Belpaire-Dethiou MC, Devogelaer JP, Van Hecke P, et al. Beneficial effects of creatine supplementation in dystrophic patients. Muscle Nerve. 2003 May. 27 (5):604-10. [QxMD MEDLINE Link].
Kley RA, Tarnopolsky MA, Vorgerd M. Creatine for treating muscle disorders. Cochrane Database Syst Rev. 2011 Feb 16. 2:CD004760. [QxMD MEDLINE Link].
Hamed SA. Drug evaluation: PTC-124--a potential treatment of cystic fibrosis and Duchenne muscular dystrophy. IDrugs. 2006 Nov. 9 (11):783-9. [QxMD MEDLINE Link].
Translarna. European Medicines Agency. Available at https://www.ema.europa.eu/en/medicines/human/EPAR/translarna. February 10, 2022; Accessed: January 24, 2023.
Zaidman C, Proud C, McDonald C, Mason S, Guridi M, Wang S, et al. One-year data from ENDEAVOR, a phase 1b trial of delandistrogene moxeparvovec (SRP-9001) in patients with Duchenne muscular dystrophy (DMD) (abstr S48.003). Neurology. 2023 Apr 25. 100 (17 Suppl 2):3717. [Full Text].
Elevidys (delandistrogene moxeparvovec) [package insert]. Cambridge, MA: Sarepta Therapeutics. June 2023. Available at [Full Text].
A gene transfer therapy study to evaluate the safety and efficacy of SRP-9001 (delandistrogene moxeparvovec) in participants with Duchenne muscular dystrophy (DMD) (EMBARK). ClinicalTrials.gov. Available at https://www.clinicaltrials.gov/study/NCT05096221. 2022 Nov 15; Accessed: June 23, 2023.
Ragot T, Vincent N, Chafey P, Vigne E, Gilgenkrantz H, Couton D, et al. Efficient adenovirus-mediated transfer of a human minidystrophin gene to skeletal muscle of mdx mice. Nature. 1993 Feb 18. 361 (6413):647-50. [QxMD MEDLINE Link].
Wang Z, Allen JM, Riddell SR, Gregorevic P, Storb R, Tapscott SJ, et al. Immunity to adeno-associated virus-mediated gene transfer in a random-bred canine model of Duchenne muscular dystrophy. Hum Gene Ther. 2007 Jan. 18 (1):18-26. [QxMD MEDLINE Link].
Howell JM, Lochmüller H, O'Hara A, Fletcher S, Kakulas BA, Massie B, et al. High-level dystrophin expression after adenovirus-mediated dystrophin minigene transfer to skeletal muscle of dystrophic dogs: prolongation of expression with immunosuppression. Hum Gene Ther. 1998 Mar 20. 9 (5):629-34. [QxMD MEDLINE Link].
Griggs RC, Karparti G, eds. Myoblast Transfer Therapy (Advances in Experimental Medicine and Biology). Dordrecht, Netherlands: Kluwer Academic Publishers; 1990.
Rando TA. Non-viral gene therapy for Duchenne muscular dystrophy: progress and challenges. Biochim Biophys Acta. 2007 Feb. 1772 (2):263-71. [QxMD MEDLINE Link].
Wells DJ. Therapeutic restoration of dystrophin expression in Duchenne muscular dystrophy. J Muscle Res Cell Motil. 2006. 27 (5-7):387-98. [QxMD MEDLINE Link].
Judge LM, Chamberlain JS. Gene therapy for Duchenne muscular dystrophy: AAV leads the way. Acta Myol. 2005 Dec. 24 (3):184-93. [QxMD MEDLINE Link].
Brooke MH, Fenichel GM, Griggs RC, Mendell JR, Moxley R, Florence J, et al. Duchenne muscular dystrophy: patterns of clinical progression and effects of supportive therapy. Neurology. 1989 Apr. 39 (4):475-81. [QxMD MEDLINE Link].
Heckmatt JZ, Dubowitz V, Hyde SA, Florence J, Gabain AC, Thompson N. Prolongation of walking in Duchenne muscular dystrophy with lightweight orthoses: review of 57 cases. Dev Med Child Neurol. 1985 Apr. 27 (2):149-54. [QxMD MEDLINE Link].
Drennan J. Neuromuscular disorders. Lowell WW, Morrissy RT, Winter RB, eds. Lovell and Winter's Pediatric Orthopaedics. 3rd ed. Philadelphia: Lippincott Williams & Wilkins; 1990. 381.
Yount CC. The role of tensor fascia femoris in certain deformities of the lower extremities. J Bone Joint Surg. 1926. 8:171-93.
Suk KS, Lee BH, Lee HM, Moon SH, Choi YC, Shin DE, et al. Functional outcomes in Duchenne muscular dystrophy scoliosis: comparison of the differences between surgical and nonsurgical treatment. J Bone Joint Surg Am. 2014 Mar 5. 96 (5):409-15. [QxMD MEDLINE Link].
Brooks JT, Sponseller PD. What's New in the Management of Neuromuscular Scoliosis. J Pediatr Orthop. 2016 Sep. 36 (6):627-33. [QxMD MEDLINE Link].
Jain A, Hassanzadeh H, Strike SA, Menga EN, Sponseller PD, Kebaish KM. Pelvic Fixation in Adult and Pediatric Spine Surgery: Historical Perspective, Indications, and Techniques: AAOS Exhibit Selection. J Bone Joint Surg Am. 2015 Sep 16. 97 (18):1521-8. [QxMD MEDLINE Link].
Gaine WJ, Lim J, Stephenson W, Galasko CS. Progression of scoliosis after spinal fusion in Duchenne's muscular dystrophy. J Bone Joint Surg Br. 2004 May. 86 (4):550-5. [QxMD MEDLINE Link].
Sengupta DK, Mehdian SH, McConnell JR, Eisenstein SM, Webb JK. Pelvic or lumbar fixation for the surgical management of scoliosis in duchenne muscular dystrophy. Spine (Phila Pa 1976). 2002 Sep 15. 27 (18):2072-9. [QxMD MEDLINE Link].
Mubarak SJ, Morin WD, Leach J. Spinal fusion in Duchenne muscular dystrophy--fixation and fusion to the sacropelvis?. J Pediatr Orthop. 1993 Nov-Dec. 13 (6):752-7. [QxMD MEDLINE Link].
Tian C, Wong B, Hornung L, et al. G.P.171: Age-specific prevalence of osteoporosis and frequency of poor bone health indices in Duchenne Muscular Dystrophy. Neuromuscul Disord. 2014 Oct. 24 (9-10):857.
Singh A, Schaeffer EK, Reilly CW. Vertebral Fractures in Duchenne Muscular Dystrophy Patients Managed With Deflazacort. J Pediatr Orthop. 2018 Jul. 38 (6):320-324. [QxMD MEDLINE Link].
Buckner JL, Bowden SA, Mahan JD. Optimizing Bone Health in Duchenne Muscular Dystrophy. Int J Endocrinol. 2015. 2015:928385. [QxMD MEDLINE Link]. [Full Text].
[Guideline] Noritz GH, Murphy NA, Neuromotor Screening Expert Panel. Motor delays: early identification and evaluation. Pediatrics. 2013 Jun. 131 (6):e2016-27. [QxMD MEDLINE Link]. [Full Text].
Muscular dystrophy: diagnostic tools. Centers for Disease Control and Prevention. Available at http://www.cdc.gov/ncbddd/musculardystrophy/diagnostic-tool.html. September 22, 2022; Accessed: January 24, 2023.
Early diagnosis makes a difference. ChildMuscleWeakness.org. Available at http://www.childmuscleweakness.org/index.php. 2023; Accessed: January 24, 2023.
[Guideline] Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010 Jan. 9 (1):77-93. [QxMD MEDLINE Link]. [Full Text].
[Guideline] Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L, et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: implementation of multidisciplinary care. Lancet Neurol. 2010 Feb. 9 (2):177-89. [QxMD MEDLINE Link]. [Full Text].
[Guideline] Birnkrant DJ, Bushby K, Bann CM, Apkon SD, Blackwell A, Brumbaugh D, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018 Mar. 17 (3):251-267. [QxMD MEDLINE Link].
[Guideline] Birnkrant DJ, Bushby K, Bann CM, Alman BA, Apkon SD, Blackwell A, et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol. 2018 Apr. 17 (4):347-361. [QxMD MEDLINE Link].
[Guideline] Birnkrant DJ, Bushby K, Bann CM, Apkon SD, Blackwell A, Colvin MK, et al. Diagnosis and management of Duchenne muscular dystrophy, part 3: primary care, emergency management, psychosocial care, and transitions of care across the lifespan. Lancet Neurol. 2018 May. 17 (5):445-455. [QxMD MEDLINE Link].
[Guideline] Birnkrant DJ, Panitch HB, Benditt JO, Boitano LJ, Carter ER, Cwik VA, et al. American College of Chest Physicians consensus statement on the respiratory and related management of patients with Duchenne muscular dystrophy undergoing anesthesia or sedation. Chest. 2007 Dec. 132 (6):1977-86. [QxMD MEDLINE Link]. [Full Text].
[Guideline] Tawil R, Kissel JT, Heatwole C, et al. Evidence-based guideline summary: Evaluation, diagnosis, and management of facioscapulohumeral muscular dystrophy: Report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology and the Practice Issues Review Panel of the American Association of Neuromuscular & Electrodiagnostic Medicine. Neurology. 2015 Jul 28. 85 (4):357-64. [QxMD MEDLINE Link]. [Full Text].
[Guideline] Wang CH, Bonnemann CG, Rutkowski A, et al, International Standard of Care Committee for Congenital Muscular Dystrophy. Consensus statement on standard of care for congenital muscular dystrophies. J Child Neurol. 2010 Dec. 25 (12):1559-81. [QxMD MEDLINE Link]. [Full Text].
[Guideline] Kang PB, Morrison L, Iannaccone ST, et al, Guideline Development Subcommittee of the American Academy of Neurology and the Practice Issues Review Panel of the American Association of Neuromuscular & Electrodiagnostic Medicine. Evidence-based guideline summary: evaluation, diagnosis, and management of congenital muscular dystrophy: Report of the Guideline Development Subcommittee of the American Academy of Neurology and the Practice Issues Review Panel of the American Association of Neuromuscular & Electrodiagnostic Medicine. Neurology. 2015 Mar 31. 84 (13):1369-78. [QxMD MEDLINE Link]. [Full Text].
[Guideline] Narayanaswami P, Weiss M, Selcen D, et al, Guideline Development Subcommittee of the American Academy of Neurology, Practice Issues Review Panel of the American Association of Neuromuscular & Electrodiagnostic Medicine. Evidence-based guideline summary: diagnosis and treatment of limb-girdle and distal dystrophies: report of the guideline development subcommittee of the American Academy of Neurology and the practice issues review panel of the American Association of Neuromuscular & Electrodiagnostic Medicine. Neurology. 2014 Oct 14. 83 (16):1453-63. [QxMD MEDLINE Link]. [Full Text].








































Comentarios